From sequence to signature: Uncovering multiscale AMR features across bacterial pathogens with supervised machine learning
From sequence to signature: Uncovering multiscale AMR features across bacterial pathogens with supervised machine learning
Ghosh, A.; Brenner, E. P.; Vang, C. K.; Wolfe, E. P.; Burke, J. T.; Lesiyon, R. L.; Manpearl, K. R.; Sridhar, V.; Krol, J. D.; Boyer, E.; Bilodeaux, J. M.; Jongnarangsin, K.; Majlessi, E. J.; Ravi, J.
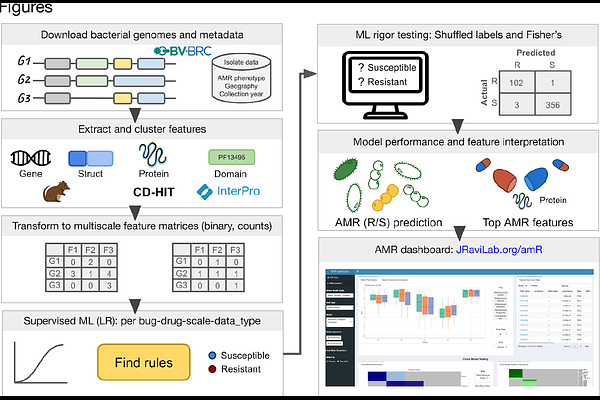
AbstractSince the clinical introduction of antibiotics in the 1940s, antimicrobial resistance (AMR) has become an increasingly dire threat to global public health. Pathogens acquire AMR much faster than we discover new drugs, warranting new methods to better understand the molecular underpinnings of AMR. Traditional approaches for detecting AMR in novel bacterial strains are time-consuming and labor-intensive. However, advances in sequencing technology offer a plethora of bacterial genome data, and computational approaches like machine learning (ML) provide an optimistic scope for in silico AMR prediction. Here, we introduce a comprehensive multiscale ML approach to predict AMR phenotypes and identify AMR molecular features associated with a single drug or drug family, stratified by time and geographical locations. As a case study, we focus on a subset of the World Health Organization\'s Bacterial Priority Pathogens, frequently drug-resistant and nosocomial bacteria: Enterococcus faecium, Staphylococcus aureus, Acinetobacter baumannii, Pseudomonas aeruginosa, and two Enterobacter species. We started with sequenced genomes with lab-derived AMR phenotypes, constructed pangenomes, clustered protein sequences, and extracted protein domains to generate pangenomic features across molecular scales. To uncover the molecular mechanisms behind drug-/drug family-specific resistance, we trained logistic regression ML models on our datasets. These yielded ranked lists of AMR-associated genes, proteins, and domains. In addition to recapitulating known AMR features, our models identified novel candidates for experimental validation. They performed well across molecular scales, data types (binary vs. counts), and drugs. The models achieved a median normalized Matthews correlation coefficient of 0.89, suggesting robust performance across molecular scales with counts or presence/absence of features. Model performance showed resilience even when evaluated on geographical and temporal holdouts. Our holistic approach promises reliable prediction of existing and developing resistance in newly sequenced pathogen genomes, along with the mechanistic molecular contributors of AMR.