Variant Classification Using Proteomics-Informed Large Language Models Increases Power of Rare Variant Association Studies and Enhances Target Discovery
Variant Classification Using Proteomics-Informed Large Language Models Increases Power of Rare Variant Association Studies and Enhances Target Discovery
Gillies, C. E.; Mbatchou, J.; Habegger, L.; Kessler, M. D.; Bao, S.; Balasubramanian, S.; Delaneau, O.; Kosmicki, J. A.; Regeneron Genetics Center, ; Willer, C. J.; Kang, H. M.; Baras, A.; Reid, J.; Marchini, J.; Abecasis, G. R.; Ghoussaini, M.
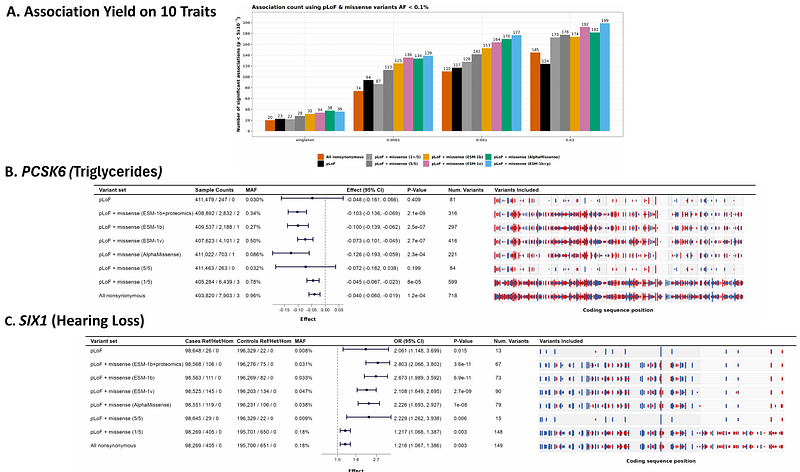
AbstractRare variant association analysis, which assesses the aggregate effect of rare damaging variants within a gene, is a powerful strategy for advancing knowledge of human biology. Numerous models have been proposed to identify damaging coding variants, with the most recent ones employing deep learning and large language models (LLM) to predict the impact of changes in coding sequences. Here, we use newly available proteomics data on 2,898 proteins across 46,665 individuals to evaluate and refine LLM predictors of damaging variants. Using one of these refined models, we evaluate association between rare damaging variants and human phenotypes at 241 positive control gene-trait pairs. Among these gene-trait pairs, our proteomics-guided model outperforms an ensemble of conventional approaches including PolyPhen2, Mutation Taster, SIFT, and LRT, as well as newer machine learning approaches for identifying damaging missense variants, such as CADD, ESM-1v, ESM-1b and AlphaMissense. When attempting to recover known associations by correctly separating damaging singleton missense variants from other singleton variants, our approach recapitulates 36.5% of gene-trait pairs with known associations, exceeding all the alternatives we considered. Furthermore, when we apply our model to 10 exemplary traits from the UK Biobank, we identify 177 gene-trait associations - again exceeding all other approaches. Our results demonstrate that summary statistics from large-scale human proteomics data enable evaluation and refinement of coding variant classification LLMs, improving discovery potential in human genetic studies.