Horse, not zebra: accounting for lineage abundance in maximum likelihood phylogenetics
Horse, not zebra: accounting for lineage abundance in maximum likelihood phylogenetics
De Maio, N.
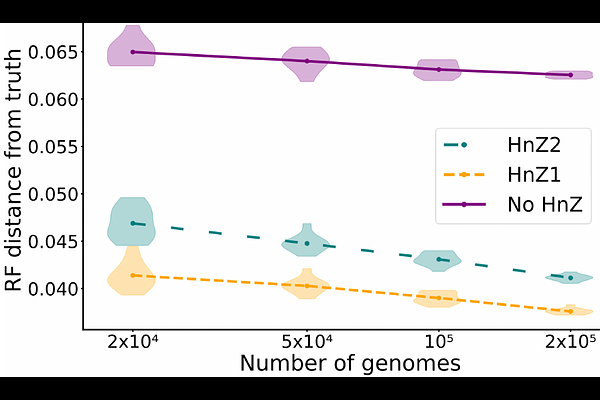
AbstractMaximum likelihood phylogenetic methods are popular approaches for estimating evolutionary histories. These methods do not assume prior hypotheses regarding the shape of the phylogenetic tree, and this lack of prior assumptions can be useful in particular in case of idiosyncratic sampling patterns. For example, the rate at which species are sequenced can differ widely between lineages, with lineages more of interest to humans being usually sequenced more often than others. However, in some settings sampling can be lineage-agnostic. In genomic epidemiology, for example, the sequencing rate can change through time or across locations, but is often agnostic to the specific pathogen strain being sequenced. In this scenario, one expects that the abundance of a pathogen strain at a specific time and location in the host population is reflected in the relative abundance of that strain among the genomes sequenced at that time and location. Here, I show that this simple assumption, when appropriate and incorporated within maximum likelihood phylogenetics, can greatly improve the accuracy of phylogenetic inference. This is similar to the famous medical principle ``when you hear hoofbeats, think of horses, not zebras''. In our application this means that, when for example observing a (possibly incomplete) genome sequence that has a similar likelihood of belonging to multiple different strains, I aim to prioritize phylogenetic placement onto a common strain (the "horse", a common disease) rather than a rare one (the "zebra", a rare disease). I introduce and assess two separate approaches to achieve this. The first approach rescales the likelihood of a phylogenetic tree by the number of distinct binary topologies obtainable by arbitrarily resolving multifurcations in the tree. This approach is based on a new interpretation of multifurcating phylogenetic trees particularly relevant at low divergence: multifurcations represent a lack of signal for resolving the bifurcating topology rather than an instantaneous multifurcating event, and so a multifurcating tree is interpreted as the set of bifurcating trees consistent with the multifurcating one, rather than as a single multifurcating topology. The second approach instead includes a tree prior that assumes that genomes are sequenced at a rate proportional to their abundance. Both approaches favor phylogenetic placement at abundant lineages, and using simulations I show that both methods dramatically improve the accuracy of phylogenetic inference in scenarios like SARS-CoV-2 phylogenetics, where large multifurcations are common. This considerable impact is also observed in real pandemic-scale SARS-CoV-2 genome data, where accounting for lineage prevalence reduces phylogenetic uncertainty by around one order of magnitude. Both approaches were implemented as part of the free and open source phylogenetic software MAPLE v0.7.5.4 https://github.com/NicolaDM/MAPLE.