iTissue
iTissue
Zambrana, C.; Malod-Dognin, N.; Przulj, N.
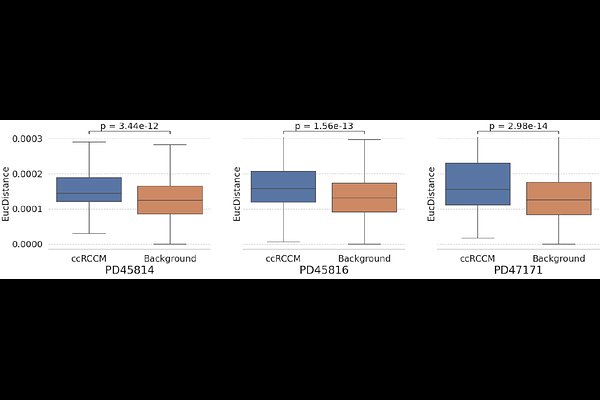
AbstractTumors are highly heterogeneous tissues, and this heterogeneity impacts metastasis and its progression. Spatial data are essential for studying these hetrogeneity. However, its limited resolution makes crucial the integration of spatial data with molecular networks to identify relevant genes for the progression of metastasis. We develop a new data integration method, named iTissue, based on Graph Non-negative Matrix Tri-Factorization that integrates spatial and single-cell transcriptomics with molecular interaction networks, producing low-dimensional space. For three clear cell renal cell carcinoma (ccRCC) patients, we applied iTissue to their tumour-core and tumour-normal interface samples. We identified the top 100 with the highest euclidean distance between these two samples, since we show that the genes with the highest euclidean distance are relevant for ccRCC. By taking the intersection of the top 100 genes for the three patients, we identify 50 genes enriched in GO-BP terms relevant for metastasis. Moreover, we find that we can cluster the three regions of the tumour-normal interface (healthy tissue, tumour tissue and interface) using the identified genes. Our framework is general and can enable insight into other diseases.