Molecular insights on the mechanism of α1-antitrypsin condensate formation and maturation
Molecular insights on the mechanism of α1-antitrypsin condensate formation and maturation
Sanchez-Burgos, I.; Tejedor, A. R.; Collepardo-Guevara, R.; de la Serna, J. B.; Espinosa, J. R.
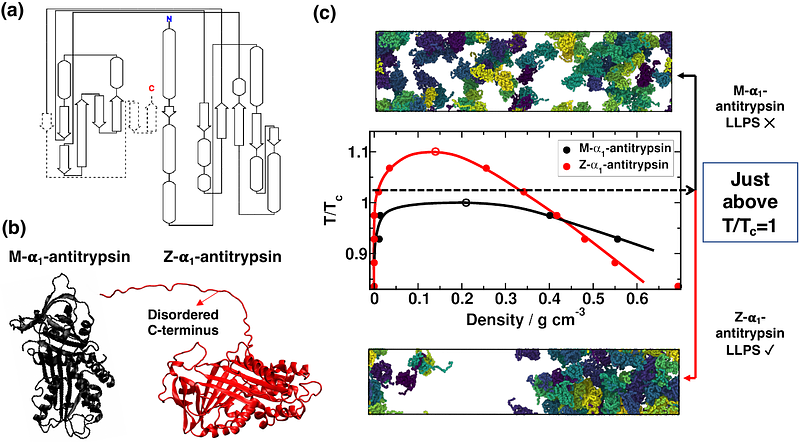
AbstractThe deficiency of 1-antitrypsin protein is a genetic disorder characterized by the accumulation of misfolded protein aggregates within hepatocytes, leading to liver dysfunction. In the lung, it is found in macrophages, bronchial and epithelial alveolar cells type 2, leading to pulmonary emphysema. Despite extensive research, the precise mechanism underlying the formation of 1-antitrypsin inclusion bodies remain elusive. In this study, we combine equilibrium and non-equilibrium molecular dynamics simulations to elucidate the intricate process of 1-antitrypsin condensate formation and maturation. Our mechanistic model explains cluster accumulation--specifically the onset of this pathogenesis--through the emergence of phase-separated liquid-like protein droplets, which subsequently undergo inter-protein {beta}-sheet transitions between misfolded variants, resulting in solid-like clusters. We find that this mechanism only applies to the misfolded variant, Z-1-antitrypsin, which phase-separates driven by its disordered C-terminus. In contrast, the native protein, M-1-antitrypsin, shows much lower propensity to phase-separate and form kinetically trapped aggregates. Furthermore, we explore how Z-1-antitrypsin exhibits an increased capacity to form condensates near external walls with different types of interactions. Such conditions can be similar to those found within the endoplasmic reticulum membrane, where phase separation and hardening take place. Overall, our results shed light on the molecular basis of 1-antitrypsin-related disorders and provide valuable microscopic insights for the development of therapeutic strategies targeting protein misfolding and aggregation-related disorders.