A neuropsychiatric disease-associated mutation in LRRC8B disrupts cellular calcium signaling, mitochondrial function, and bioenergetics
A neuropsychiatric disease-associated mutation in LRRC8B disrupts cellular calcium signaling, mitochondrial function, and bioenergetics
Ajith, A.; S, D. S.; Sharma, R.; Ghosh, A. K.; Bera, A. K.
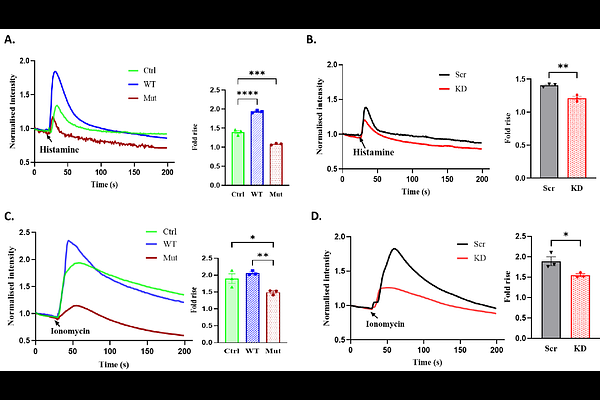
AbstractLeucine-rich repeat-containing 8 (LRRC8) proteins form the volume-regulated anion channel (VRAC) and participate in diverse physiological processes, including cell volume regulation, gliotransmitter release, and insulin secretion. In mammals, five paralogs (LRRC8A to E) exist; LRRC8A is the obligatory subunit that assembles into functional hexameric channels with LRRC8C, D, or E. LRRC8B is distinct: we previously demonstrated its role in regulating endoplasmic reticulum (ER) Ca2+ homeostasis and ER Ca2+; leak. A LRRC8B variant (Y380S) identified in an Indian family with severe mental illness has been associated with disease pathology, but its molecular and cellular consequences remain unknown. Here, we show that this disease-associated mutant perturbs Ca2+ signalling, mitochondrial bioenergetics, and redox homeostasis. Both wild-type and mutant LRRC8B localize to the ER and mitochondria. LRRC8B knockdown significantly reduced mitochondrial Ca2+ uptake and maximal respiratory the Y380S mutant phenocopied LRRC8B knockdown, altering ER Ca2+ release, elevating basal cytosolic Ca2+ , and impairing mitochondrial Ca2+ uptake, consistent with a dominant-negative mechanism. The mutant further induced mitochondrial dysfunction, including loss of membrane potential, oxidative stress, and defective antioxidant responses, ultimately compromising cellular bioenergetics and viability. Mechanistically, the Y380S mutation disrupted LRRC8B interaction with the mitochondrial outer membrane channel VDAC. These findings identify LRRC8B and VDAC coupling as a key determinant of mitochondrial Ca2+ handling and provide a mechanistic link between LRRC8B dysfunction and neuropsychiatric disease