PAH DEFICIENT PATHOLOGY IN HUMANIZED c.1066-11G>A PHENYLKETONURIA MICE
PAH DEFICIENT PATHOLOGY IN HUMANIZED c.1066-11G>A PHENYLKETONURIA MICE
Martinez-Pizarro, A.; Pico, S.; Lopez-Marquez, A.; Rodriguez-Lopez, C.; Montalvo, E.; Alvarez, M.; Castro, M.; Ramon-Maiques, S.; Perez, B.; Lucas, J. J.; Richard, E. M.; Desviat, L. R.
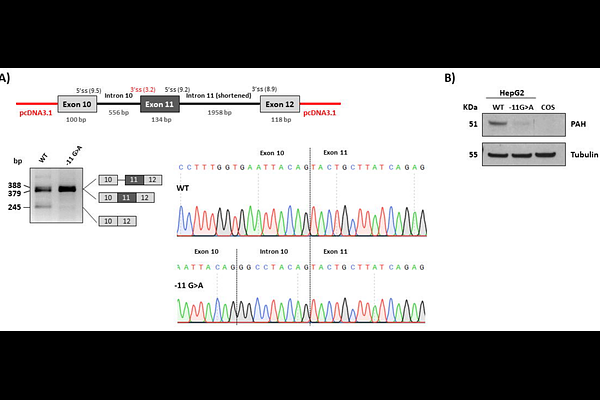
AbstractWe have generated using CRISPR/Cas9 technology a partially humanized mouse model of the neurometabolic disease phenylketonuria (PKU) carrying the prevalent splice intronic variant PAH c.1066-11G>A. This variant creates a strong alternative splice accpetor site, leading to the inclusion of 9 nucleotides coding for a 3 amino acid insertion between Q355 and Y356 of the phenylalanine hydroxylase (PAH) protein. Homozygous Pah c.1066-11A mice, with a partially humanized intron 10 sequence with the variant, accurately recapitulate the splicing defect and present almost undetectable hepatic PAH protein or activity. They exhibit fur hypopigmentation, lower brain and body weight and reduced survival. Blood and brain phenylalanine (Phe) levels are elevated, along with decreased tyrosine (Tyr), tryptophan (Trp) and monoamine neurotransmitters levels. Pah c.1066-11A mice present behavioral deficits, mainly hypoactivity and diminished social interaction, as well as locomotor deficiencies. Abnormal hind-limb clasping reflex and microophtalmia are observed. Immunohistochemistry analyses of GFAP and Iba1 markers revealed changes in the morphology of glial cells and a significant increase in GFAP and Iba1 staining signals. Western blot analyses in mouse liver lysates and in gene edited HepG2 cells with the variant showed reduced levels of chaperones DNAJC12 and HSC70/HSP70 and an increase in autophagy markers LAMP1 and LC3BII, suggesting that the mutant pQ355_Y356insGLQ PAH protein could aggregate complexed with DNAJC12 and HSP70 proteins, with the aggregates being processed by autophagy. This humanized model corresponding to a severe, classical PKU phenotype represents a useful tool for pathophysiology research in PKU and for novel therapies development.