Comparative genome analysis of Corynebacterium ulcerans Japanese isolates revealed domestically and globally diverse geographical distribution of the organism of different types.
Comparative genome analysis of Corynebacterium ulcerans Japanese isolates revealed domestically and globally diverse geographical distribution of the organism of different types.
Kimura, M.; Tsuyoshi, S.; Kuroda, M.; Senoh, M.; Takahashi, M.; Yamamoto, A.; Iwaki, M.
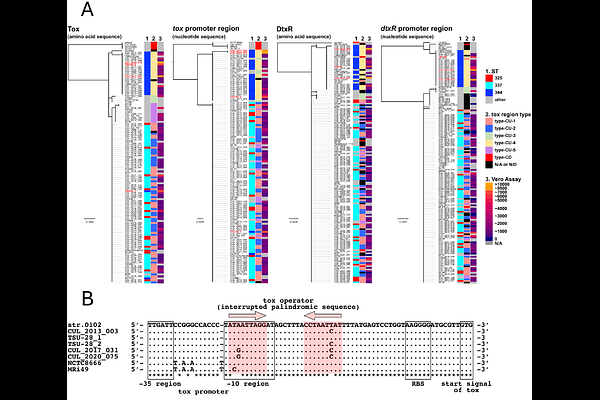
AbstractCorynebacterium ulcerans is a zoonotic pathogen causing infections in humans that are clinically indistinguishable from those of Corynebacterium diphtheriae. In many developed countries, companion animals such as dogs and cats, are the predominant sources of infection by C. ulcerans, as is the case in Japan. To date, we have collected 106 Japanese clinical and domestic animal isolates of C. ulcerans and a recently branched species, C. ramonii, most of which were isolated from humans and cats. In the present study, we performed a comparative analysis of whole-genome sequences from Japanese isolates and 597 published isolates from Japan and outside Japan. The Japanese isolates were divided into two distinct lineages: C. ulcerans and C. ramonii. The MLST types of C. ulcerans Japanese isolates exhibited a unique distribution pattern, with one major type (ST337) accounting for 69.0%, which is very rare in Europe. In addition to Japan, distinct MLST pattern compositions were observed across geographically distinct regions. The sequence types were associated with lysogenized prophage types that encode the diphtheria toxin (tox) gene and were partially associated with toxin production under certain conditions. Through SNV analysis, transmission from animals to humans has been suggested in some clinical cases.