Mass spectrometry-based quantification of proteins and post-translational modifications in dried blood: longitudinal sampling of patients with sepsis in Tanzania
Mass spectrometry-based quantification of proteins and post-translational modifications in dried blood: longitudinal sampling of patients with sepsis in Tanzania
Foster, M. W.; McMahon, T. J.; Ngocho, J. S.; Kipengele, A. H.; Violette, M.; Chen, Y.; Madut, D. B.; Plumb, R. S.; Wong, A. I.; Chen, L.; Lee, G. M.; Sakasaka, P. A.; Mmbaga, B. T.; Crump, J. A.; McClain, M. T.; Woods, C. W.; Maro, V. P.; Rubach, M. P.
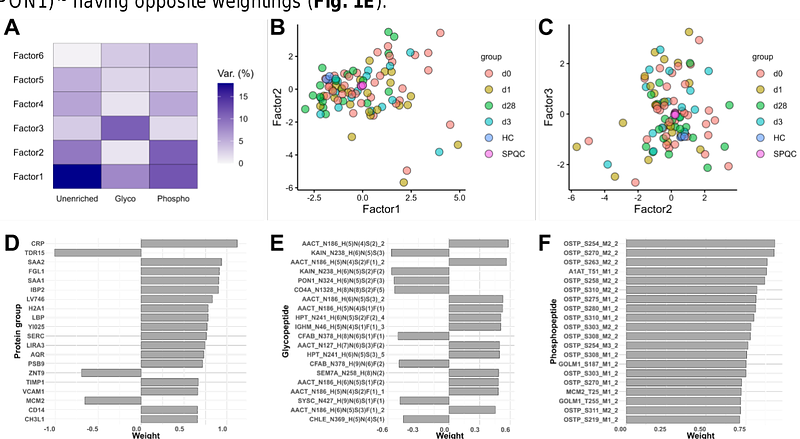
AbstractThe proteomic analysis of blood is routine for disease phenotyping and biomarker development. Whole blood is commonly separated into soluble and cellular fractions. However, this can introduce pre-analytical variability; and analysis of a single component (which is common) may ignore important pathophysiology. We have recently developed methods for the facile processing of dried blood for mass spectrometry-based quantification of the proteome, N-glycoproteome and phosphoproteome. Here, we applied this approach to 38 patients in Tanzania who presented to the hospital with sepsis. Blood was collected on Mitra devices at presentation and 1, 3 and 28-42 days post-enrollment. Processing of 96 total samples was performed in plate-based formats and completed within two days. Approximately 2,000 protein groups and 8,000 post-translational modifications were quantified in 3 LC-MS/MS runs at ~1.5 hours per sample. Analysis of differential abundance revealed blood proteome signatures of acute phase response and neutrophilic inflammation that partially resolved at the 28-42 day timepoint. Numerous analytes correlated with clinical laboratory values for c-reactive protein and white blood cell counts, as well as the Universal Vital Assessment illness severity score. These datasets serve as proof-of-concept for large scale MS-based (sub)phenotyping of disease using dried blood and are available via the ProteomeXchange consortium (PXD060377).