Best practices for improving alignment and variant calling on human sex chromosomes
Best practices for improving alignment and variant calling on human sex chromosomes
Oill, A. M. T.; Plaisier, S. B.; Phung, T. N.; Wilson, M. A.
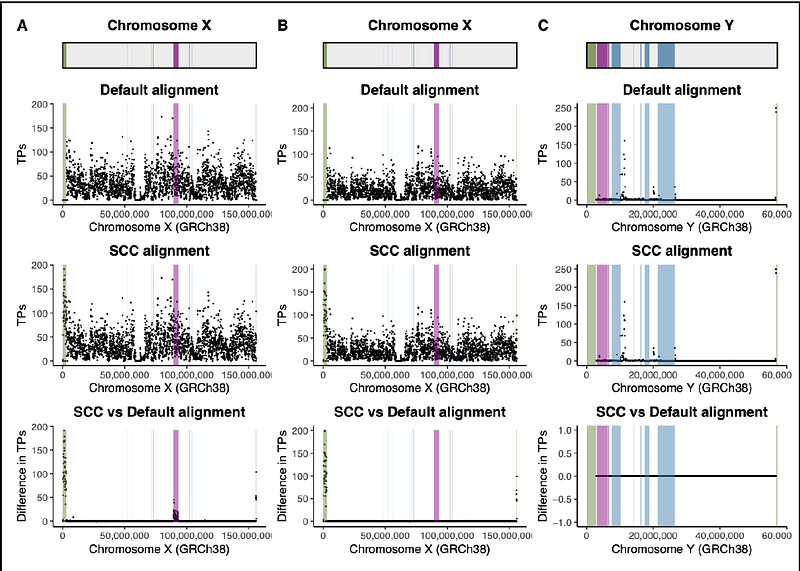
AbstractSex chromosome complement is the largest karyotypic variation observed in humans. X and Y chromosomes were once a pair of homologous autosomes. Although chromosome X and Y differentiated from one another, they still share high levels of sequence similarity in some regions, like the pseudoautosomal regions (PARs) and the X-transposed region (XTR). The sex chromosomes violate some assumptions of autosomal pairs, but are not always processed separately in genomics analyses. Here, we undertook a simulation study to assess the effects of standard autosomal versus sex chromosome complement-informed alignment, variant calling, and variant filtering strategies on variants detected on the human sex chromosomes. We find that aligning samples to a reference genome informed by the sex chromosome complement of the sample increases the number of true positives called in the PARs, and, in XX-samples only, also the XTR. In contrast, in XY-samples, masking the XTR during alignment results in a ten-fold higher rate of false positives. We further find that haploid calling on the sex chromosomes in XY-samples reduces the number of false positives compared to diploid calling, but does not decrease the number of false negatives. Improving the accuracy of variant calling results in detection of variants that could be relevant to studies of health and disease, including variants we recovered in genes implicated in cardiomyopathy, immunodeficiency, and Alzheirmer's disease, among others. We recommend future genomic analyses implement the following best practices for detecting variants: aligning samples to versions of the human reference genome informed by the sex chromosome complement of the sample and using accurate ploidy parameters when calling variants.