Host species background, defence systems, and phage tail gene architecture shape phage infectivity in cystic fibrosis-associated Achromobacter
Host species background, defence systems, and phage tail gene architecture shape phage infectivity in cystic fibrosis-associated Achromobacter
Tarasenko, A.; Papudeshi, B.; Nyugen, V.; Grigson, S. R.; Bouras, G.; Mallawaarachchi, V.; Hutton, A. L. K.; Green, R.; Ramsay, J.; Hajama, H.; Cobian Güemes, A. G.; Segall, A. M.; Warner, M. S.; Giles, S. K.; Harker, C. M.; Edwards, R. A.
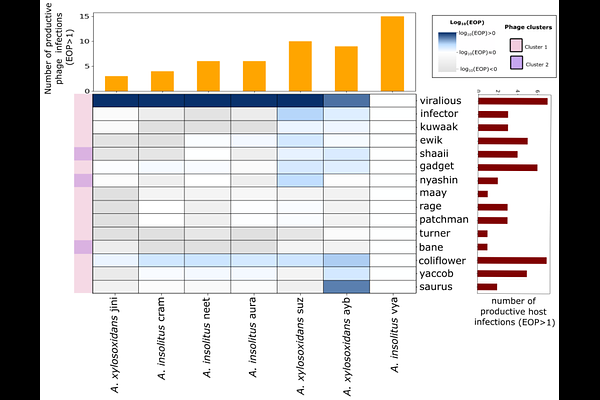
AbstractAchromobacter species are emerging multidrug-resistant (MDR) pathogens in people with cystic fibrosis. Their increasing resistance has grown an interest in phage therapy as an alternative treatment strategy. However, the factors governing phage susceptibility remain poorly understood, thereby limiting the rational selection of phage candidates. Using 15 strictly lytic Achromobacter phages and 7 clinical cystic fibrosis isolates representing Achromobacter insolitus and Achromobacter xylosoxidans, we demonstrate substantial variation in infection efficiency across all 105 phage-host combinations, variation that could not be discerned from qualitative plaque assays alone. We integrated complete bacterial and phage genomes with quantitative efficiency-of-plating (EOP) assays and lineage-aware Bayesian mixed-effects modelling to show that phage infectivity in Achromobacter is governed predominantly by bacterial lineage and strain identity, accounting for 90% of total variance in log-normalised EOP, with individual strains varying substantially in permissiveness irrespective of species membership. After accounting for this lineage structure, no individual defence system, antimicrobial resistance gene class, or phage tail cluster retained a statistically significant independent or interaction association with infectivity. Together, these findings demonstrate that bacterial strain identity is the primary driver of infection outcome. Host defence systems and phage tail-associated genes remain biologically plausible contributors; their independent effect could not be resolved after accounting for lineage structure, indicating that infection outcomes are largely strain-dependent. This work shifts the question from which individual traits predict infection to how strain lineage and specific host-phage combinations jointly determine infectivity, and argues that quantitative phenotyping of individual phage-host pairs is essential for guiding phage candidate selection and supporting rational cocktail design against multidrug-resistant Achromobacter infections in cystic fibrosis. Impact statementChronic Achromobacter infections in cystic fibrosis are increasingly difficult to treat due to multidrug resistance and biofilm formation. Although phage therapy is a promising alternative, its development is limited by poorly understood and highly variable infectivity. Here, we show that infectivity within a phage host range spans a broad quantitative continuum spanning several orders of magnitude that cannot be captured by qualitative plaque assays. These infection efficiencies are primarily structured by bacterial lineage and strain identity, while the contributions of individual genomic features remain unresolved, given the current sample size. This work provides a framework for predicting phage-host compatibility and supports a shift from empirical screening toward rational, evidence-based phage selection for MDR Achromobacter infections.